Detect the spatial context of each cell based on its neighborhood
Source:R/detectSpatialContext.R
detectSpatialContext.RdFunction to detect the spatial context (SC) of each cell. Based on its sorted (high-to-low) cellular neighborhood (CN) fractions in a spatial interaction graph, the SC of each cell is assigned as the set of CNs that cumulatively exceed a user-defined fraction threshold.
The term was coined by Bhate S. et al., Tissue schematics map the specialization of immune tissue motifs and their appropriation by tumors, Cell Systems, 2022 and describes tissue regions in which distinct CNs may be interacting.
detectSpatialContext(
object,
entry = "aggregatedNeighbors",
threshold = 0.9,
name = "spatial_context"
)Arguments
- object
a
SingleCellExperimentorSpatialExperimentobject- entry
single character specifying the
colData(object)entry containing theaggregateNeighborsDataFrame output. Defaults to "aggregatedNeighbors".- threshold
single numeric between 0 and 1 that specifies the fraction threshold for SC assignment. Defaults to 0.9.
- name
single character specifying the name of the output saved in
colData(object). Defaults to "spatial_context".
Value
returns an object of class(object) containing a new column
entry to colData(object)[[name]]
Spatial context background
The function relies on CN fractions for each cell in a spatial interaction graph (originally a k-nearest neighbor (KNN) graph).
We can retrieve the CN fractions using the buildSpatialGraph and aggregateNeighbors functions.
The window size (k for KNN) for buildSpatialGraph should reflect a length scale on which biological signals can be exchanged and depends, among others, on cell density and tissue area. In view of their divergent functionality, we recommend to use a larger window size for SC (interaction between local processes) than for CN (local processes) detection.
Subsequently, the CN fractions are sorted from high-to-low and the SC of each cell is assigned the minimal combination of SCs that additively surpass a user-defined threshold. The default threshold of 0.9 aims to represent the dominant CNs, hence the most prevalent signals, in a given window.
For more details, please refer to: Bhate S. et al., Tissue schematics map the specialization of immune tissue motifs and their appropriation by tumors, Cell Systems, 2022.
See also
filterSpatialContext for the function to filter
spatial contexts
plotSpatialContext for the function to plot
spatial context graphs
Examples
set.seed(22)
library(cytomapper)
data(pancreasSCE)
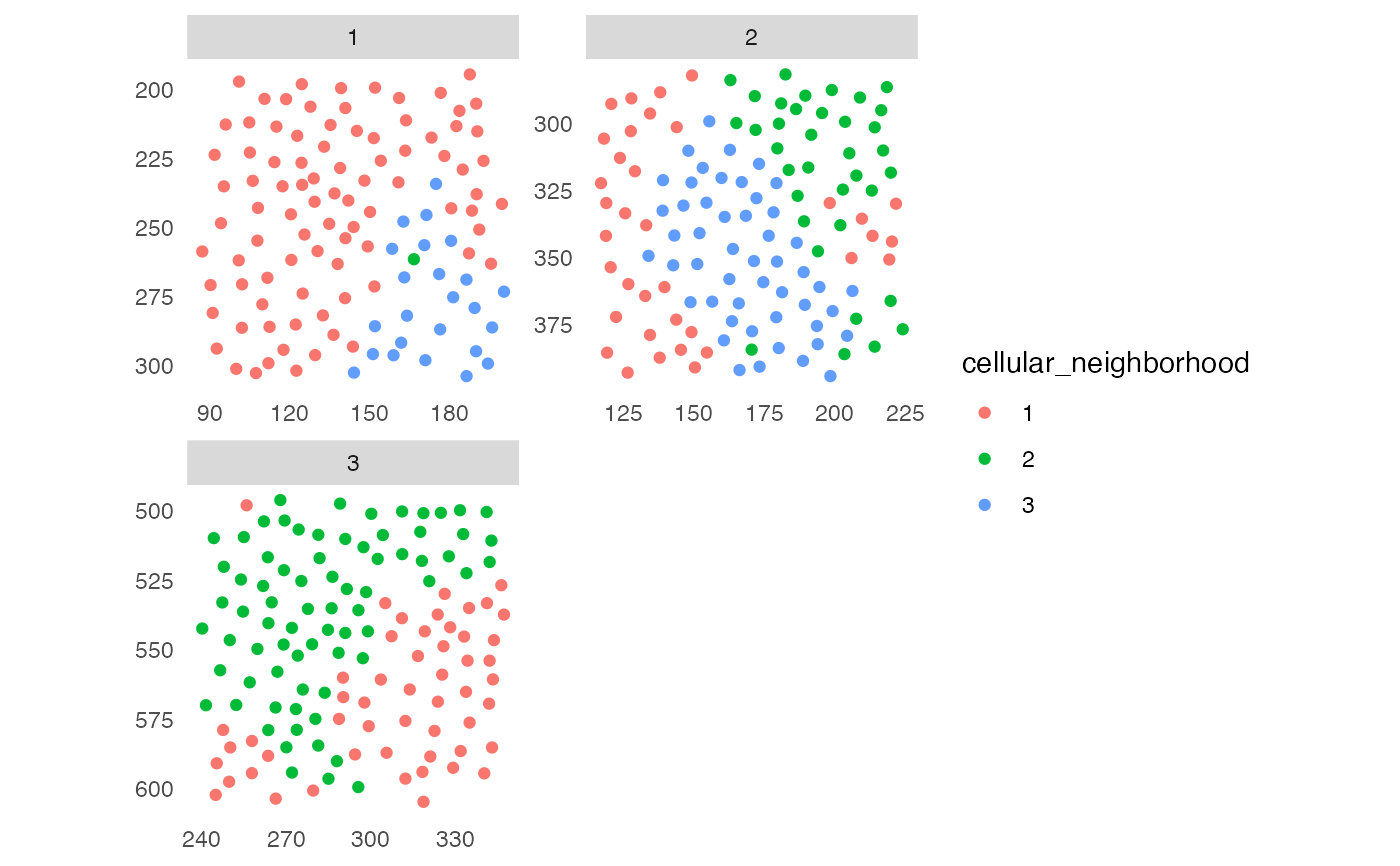
## 1. Cellular neighborhood (CN)
sce <- buildSpatialGraph(pancreasSCE, img_id = "ImageNb",
type = "knn",
name = "knn_cn_graph",
k = 5)
#> The returned object is ordered by the 'ImageNb' entry.
sce <- aggregateNeighbors(sce, colPairName = "knn_cn_graph",
aggregate_by = "metadata",
count_by = "CellType",
name = "aggregatedCellTypes")
cur_cluster <- kmeans(sce$aggregatedCellTypes, centers = 3)
sce$cellular_neighborhood <- factor(cur_cluster$cluster)
plotSpatial(sce, img_id = "ImageNb",
colPairName = "knn_cn_graph",
node_color_by = "cellular_neighborhood",
scales = "free")
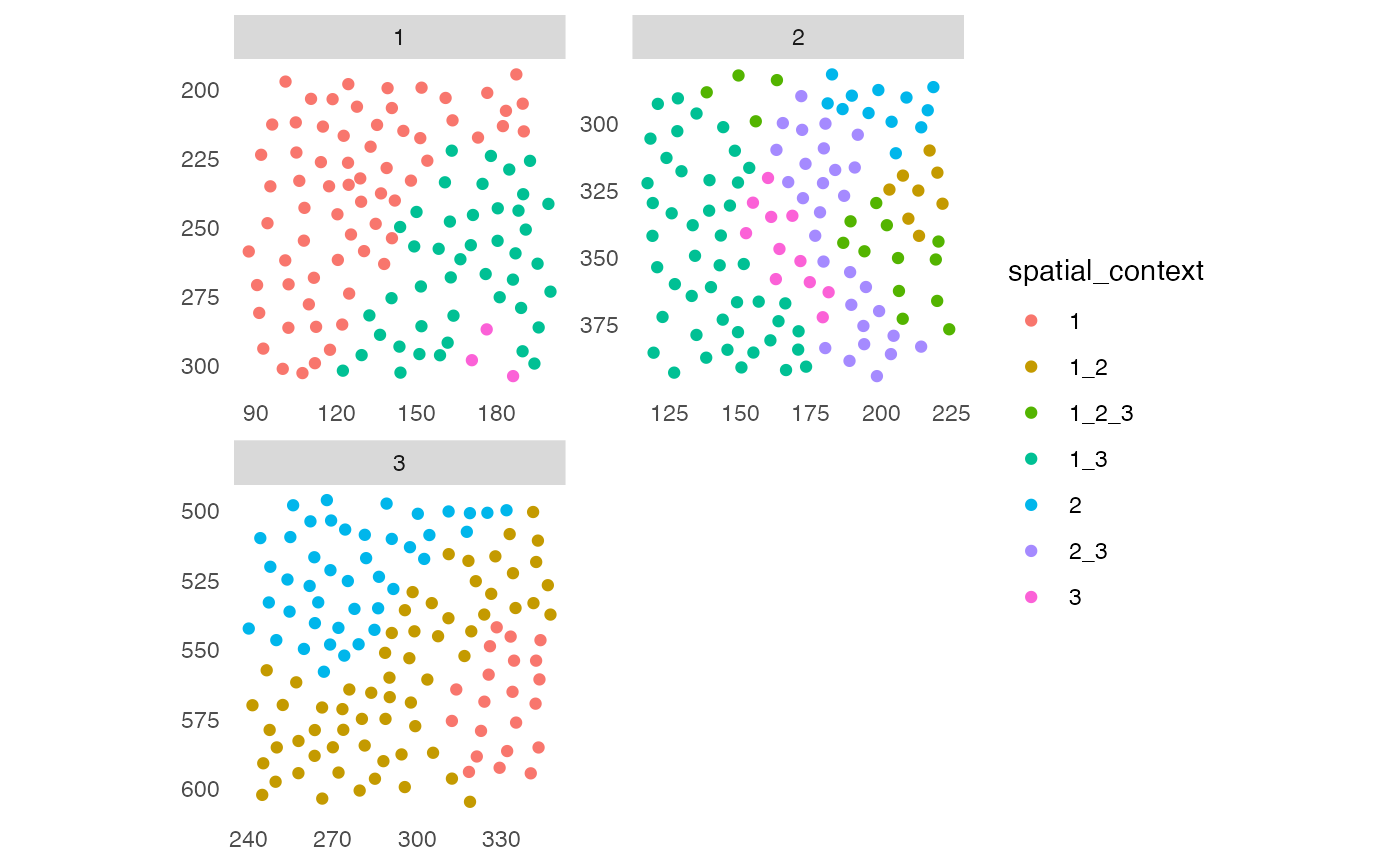
 ## 2. Spatial context (SC)
sce <- buildSpatialGraph(sce, img_id = "ImageNb",
type = "knn",
name = "knn_sc_graph",
k = 15)
#> The returned object is ordered by the 'ImageNb' entry.
sce <- aggregateNeighbors(sce, colPairName = "knn_sc_graph",
aggregate_by = "metadata",
count_by = "cellular_neighborhood",
name = "aggregatedNeighborhood")
# Detect spatial context
sce <- detectSpatialContext(sce, entry = "aggregatedNeighborhood",
threshold = 0.9)
plotSpatial(sce, img_id = "ImageNb",
colPairName = "knn_sc_graph",
node_color_by = "spatial_context",
scales = "free")
## 2. Spatial context (SC)
sce <- buildSpatialGraph(sce, img_id = "ImageNb",
type = "knn",
name = "knn_sc_graph",
k = 15)
#> The returned object is ordered by the 'ImageNb' entry.
sce <- aggregateNeighbors(sce, colPairName = "knn_sc_graph",
aggregate_by = "metadata",
count_by = "cellular_neighborhood",
name = "aggregatedNeighborhood")
# Detect spatial context
sce <- detectSpatialContext(sce, entry = "aggregatedNeighborhood",
threshold = 0.9)
plotSpatial(sce, img_id = "ImageNb",
colPairName = "knn_sc_graph",
node_color_by = "spatial_context",
scales = "free")